Our laboratory’s focus is the pathogenesis and pathophysiologic mechanisms of the chronic kidney disease-bone and mineral disorder (CKD-MBD). Kidney diseases produce a complex set of body-wide complications. Some of these contribute to the mortality of kidney diseases, which is not due to the disease itself but largely due to associated cardiovascular disease. Cardiovascular disease risk in CKD is contributed to by non-traditional risk factors which are components of the CKD-MBD syndrome.
We have discovered that renal injury and chronic kidney disease directly impair vascular, cardiac and skeletal function by production of factors involved in renal repair that are released into the circulation and contribute to the pathogenesis of CKD-MBD. The first factors we discovered to be involved in this new paradigm were the Wnt inhibitors, represented in published studies by Dkk1 and sclerostin. Wnt inhibitors are circulating factors produced by activation of renal Wnts during repair and renal fibrosis. We have shown that Dkk1 stimulates loss of osteoblast function and decreased bone formation, with less of an inhibition of bone resorption. This establishes excess osteoclast activity which persists as the initial low turnover state is converted to a high turnover state by parathyroid hormone as CKD progresses. Dkk1 also contributes to vascular disease in CKD through contributing to osteoblastic transition of cells in the arterial media and atherosclerotic plaques. Osteoblastic transition results in expression of the osteoblast transcriptome in arterial walls driven by Runx2, osterix, ATF4 and Msx2 transcription factors leading to vascular calcification. The osteoblastic transcriptome includes tissue non-specific alkaline phosphatase which metabolizes vascular pyrophosphate to Pi and increases Ca – Pi crystal formation – vascular calcification. We and others have also shown, that Pi (hyperphosphatemia) directly stimulates vascular osteoblastic function.
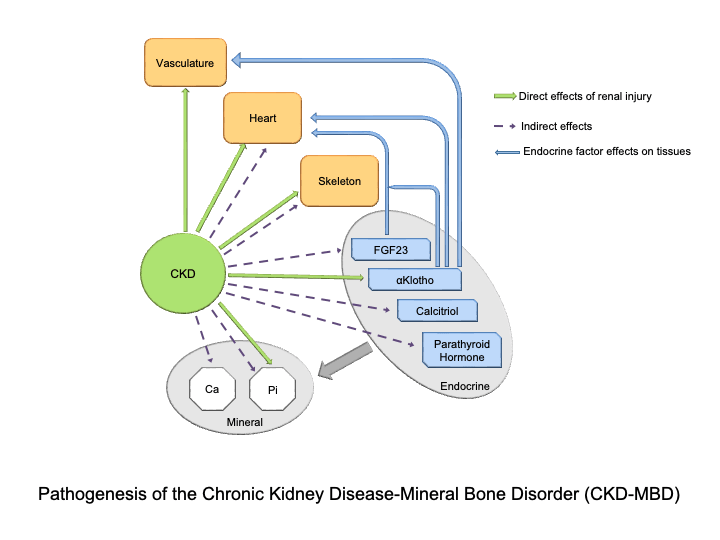
Because of the interaction of Wnt and TGFβ signaling in vascular development and vasculopathies, the second group of factors involved in renal repair/disease we studied were TGFβ superfamily members that produce signal transduction by smads and are involved in the mechanism of Wnt inhibitor function in CKD and might be potential factors affected by CKD. We already had a long history of investigation related to a key renal morphogen of the family, bone morphogenetic protein -7 (BMP-7) in renal osteodystrophy, vascular calcification and renal fibrosis. To investigate the role of the TGFβ superfamily in CKD-MBD pathogenesis, we chose to use ligand traps comprised of recombinant extracellular receptor domains fused to Fc components of immunoglobulins for the type 2 receptors. We began with a ligand trap for the murine activin receptor type IIA, ActRIIA-Fc. We first showed that ActRIIA-Fc decreased CKD stimulated vascular calcification in our model of diabetic atherosclerosis, the ldlr-/- high fat fed mouse with ablative CKD. Then we showed that the ActRIIA-Fc stimulated bone formation and inhibited bone resorption in the ldlr-/- high fat fed CKD mouse model. In a spontaneous kidney disease model, we found that ActRIIA activity was stimulated by CKD in the vasculature, skeleton, heart and kidneys of 200 day old Alport syndrome mice, and that its inhibition decreased vascular calcification, vascular osteoblastic transition, renal osteodystrophy, cardiac hypertrophy and renal fibrosis delaying progression of CKD. In each of the models of CKD that we have studied to date, we have observed stimulation of cardiac hypertrophy by CKD. This may have been related to vascular stiffness produced by vascular calcification, and this appears to be possible in the ldlr-/- high fat fed CKD mice. However, our Alport mice do not have vascular stiffness when they develop CKD and progress to kidney failure. Our hypothesis is that kidney disease stimulated repair/disease factors directly stimulate cardiac myocyte hypertrophy in CKD, and this is the project currently focused on in the laboratory.
Projects
Vascular calcification: Mechanism of stimulation by CKD
- Effects of CKD on vascular smooth cell phenotype and vascular calcification
CKD-MBD (renal osteodystrophy/osteoporosis)
- Pathogenesis of the CKD-MBD
- The effects of of CKD on osteoblast function
- Relationship of vascular calcification and bone formation in CKD
Mechanisms of BMP-7 therapy for vascular calcification
- Will BMP-7 treat rather than prevent vascular calcification? (see Figure 1)
- Will BMP-7 increase GFR in severe kidney failure?
New therapies for chronic kidney disease
- Role of Wnt inhibitor neutralization
- Role of activin inhibition
- Effects of BMP-7 therapy on renal osteodystrophy
Osteoclast biology
- Role of microRNA in osteoclastogenesis